Molecular phylogenetics is a branch of phylogenetics aimed at tracing evolutionary relationships between organisms at higher taxonomic ranks. This discipline analyzes the variation between nucleotide/amino acid sequences with a common origin and thus understands the evolution of taxa at higher levels.
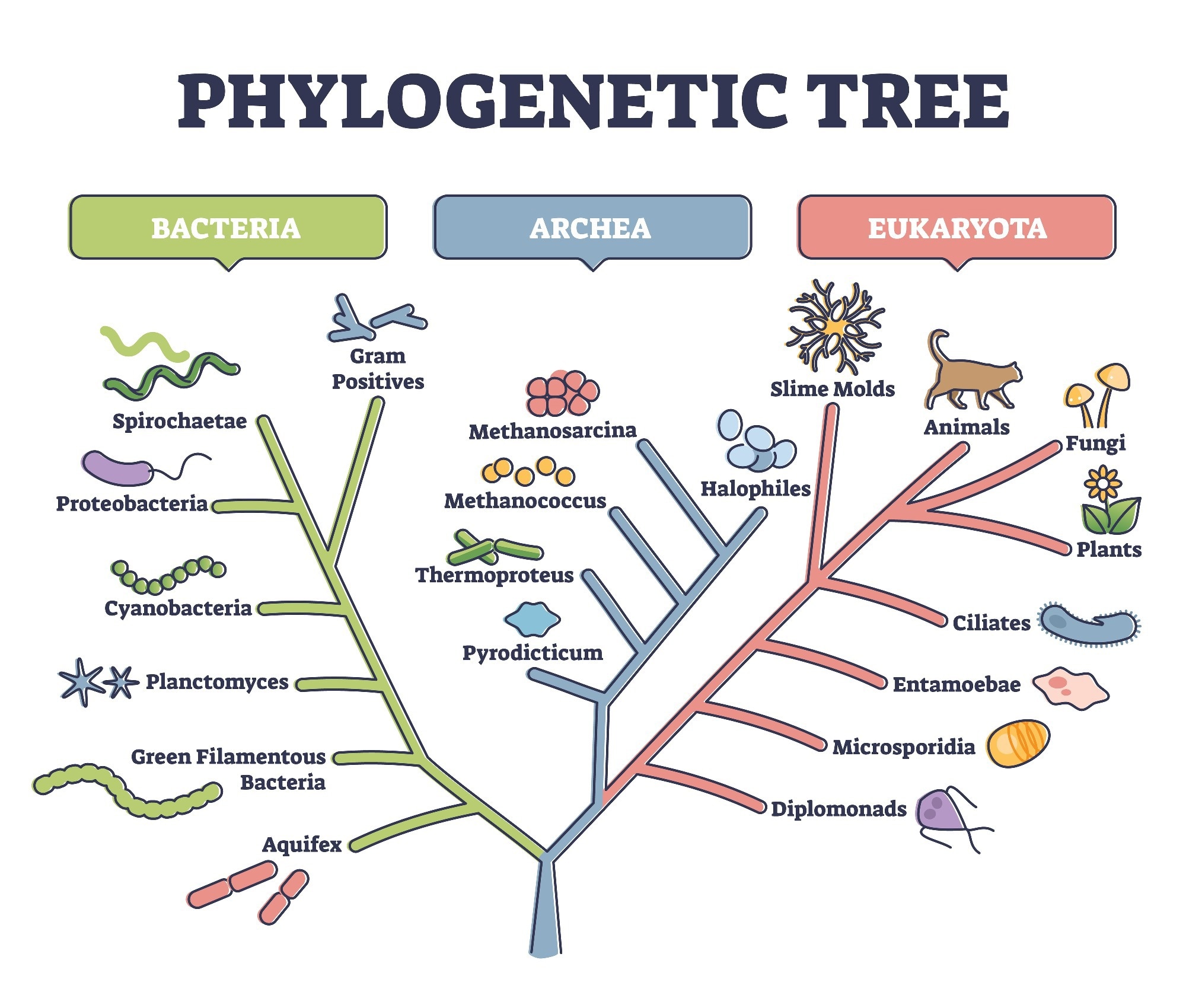
Image Credit: VectorMine/Shutterstock.com
Moreover, phylogeography is another related area of phylogenetics that studies principles, methodologies, and guidelines for the analysis of molecular variation within minor taxonomic ranks (i.e., genus and species level), which can be used to understand the biogeographic history of groups of organisms.
Molecular phylogenetics has become an integral part of molecular systematics and evolutionary biology. This discipline analyzes nucleic acids' sequence homology (DNA or RNA) or proteins to determine evolutionary links between species. Over the last few years, molecular phylogenetics has undergone deep transformations due to the emergence of high-throughput next-generation sequencing technologies and bioinformatic resources for analyzing massive genome data.
The Importance of Molecular Phylogenetics in Evolutionary Biology
Phylogenetics refers to the study of relationships between genetically related groups of organisms that form a taxonomic group or taxon. However, phylogenetic analyses are used to understand evolutionary relationships and help us determine the spread of infectious agents, explore biogeographic processes, identify incongruent cultural histories, detect genetic recombination in multiple loci, etc.
Different types of empirical biological data from the fossil record, comparative anatomy, embryology, etc., may provide useful information about common ancestry. Molecular phylogenetics is also a useful strategy that uses the linear order of nucleotides or amino acids (i.e., sequence homology) as raw material to infer common ancestry. The evolutionary relationships recovered by molecular phylogenetics are generally based on the coalescent model, a model beyond the traditional population genetics approach of neutral evolution aimed at describing the properties of gene trees.
Traditional phylogenetic trees were originally based on morphotypes/morphological characteristics, which do not always show evolutionary relationships. Over the last decades, molecular phylogenetics was gaining ground in evolutionary biology. The most widely extended molecular phylogenetic approaches have been based on the alignment of sequences (nucleotides) obtained from Sanger sequencing runs of PCR products.
Nowadays, the advent of modern next-sequencing platforms, as well as the growing number of publicly available sequencing datasets, is enabling the development of new phylogenetic analyses based on the comparison of non-coding genome regions, including highly repetitive telomeric/Transposable Elements rich regions. These non-coding sequences can play important roles in genome evolution and organization.
Moreover, the design of complex bioinformatic tools has also allowed the reconstruction of evolutionary links by comparing large portions of closely related genomes, a relatively new approach known as phylogenomics.
Current Methods in Molecular Phylogenetics
Molecular phylogenetics can be used to estimate divergence times, test hypotheses, identify signals of positive selection in large genome-scale datasets, etc. There are different algorithms for estimating phylogenetic relationships; some of the most widely extended include Parsimony, Distance-based methods, Maximum Likelihood, and Bayesian inference.
- Maximum parsimony is likely the most frequently applied phylogenetic method for inferring the origin and evolution of molecular sequences. The evolutionary trees estimated by Maximum parsimony take into consideration the fewest number of steps to generate the observed variation from common ancestral sequences. A maximum parsimony tree is a consensus of parsimonious trees deduced by minimizing the number of nucleotide/amino acid substitutions required to build the tree.
- Maximum Likelihood is a powerful approach for estimating the parameters of a probability model, and it is also widely used for inferring phylogenetic trees from sequence data. The maximum likelihood criterion is a useful strategy to estimate the evolutionary history of a taxonomic group by assessing the probabilities for a proposed set of parameters (i.e., the 'molecular model') to give rise to the observed dataset.
- Bayesian statistics is based on Bayes' theorem to estimate the probabilities for a given hypothesis as more evidence becomes available. In molecular phylogenetics, Bayesian statistics can infer the posterior probability of an evolutionary event based on prior probability distributions incorporated into assessing a set of sequence data.

Image Credit: BB DESIGN STOCK/Shutterstock.com
What Does the Future of Molecular Phylogenetics Look Like?
Recent advances in sequencing technologies and the development of new software and algorithms for inferring evolutionary trees (phylogenies) face a promising future in molecular phylogenetics. It is important to note that several technical difficulties (such as library preparation), a yet predominant focus on non-model organisms, and expensive high-throughput DNA sequencing data are some major issues that still need to be addressed in this field.
Developing powerful phylogeny packages that allow the simultaneous analysis of hundreds of loci is likely the next step for accurate genetic measurements in molecular phylogeography and phylogenetics. In this sense, it is important to highlight that probabilistic Bayesian estimations are gaining ground for examining evolutionary relationships at higher taxonomic levels.
Sources:
- Fabreti, Luiza Guimarães, and Sebastian Höhna. "Bayesian inference of phylogeny is robust to substitution model over-parameterization." bioRxiv (2022).
- McCormack, John E., et al. "Applications of next-generation sequencing to phylogeography and phylogenetics." Molecular Phylogenetics and Evolution 66.2 (2013): 526-538.
- Meyer, Xavier, et al. "Simultaneous Bayesian inference of phylogeny and molecular coevolution." Proceedings of the National Academy of Sciences 116.11 (2019): 5027-5036.
- Puttick, Mark N., et al. "Probabilistic methods outperform parsimony in the phylogenetic analysis of data simulated without a probabilistic model." Palaeontology 62.1 (2019): 1-17.
- Whelan, Simon, Pietro Liò, and Nick Goldman. "Molecular phylogenetics: state-of-the-art methods for looking into the past." Trends in Genetics 17.5 (2001): 262-272.
- Yang, Ziheng, and Bruce Rannala. "Molecular phylogenetics: principles and practice." Nature Reviews Genetics 13.5 (2012): 303-314.
Further Reading